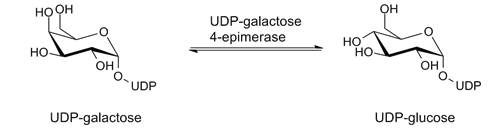
La galactosemia es una enfermedad caracterizada por la incapacidad de metabolizar la galactosa en glucosa. Ésto se debe a que el sujeto hereda un gen defectuoso de cada progenitor (enfermedad autosómica recesiva). La galactosa es un monosacárido obtenido principalmente de la hidrólisis de la lactosa contenida en la leche, aunque también puede estar presente en otros alimentos. La galactosa se absorbe en el intestino y principalmente se transforma en glucosa en el hígado.
Existe una deficiencia en la enzima galactosa-I-fostatasa uridil-transferasa, que es imprescindible para pasar de galactosa a glucosa. Normalmente cuando una persona consume un producto que contiene lactosa, el metabolismo degrada la lactosa en glucosa y galactosa. Una cantidad excesiva de galactosa en sangre causa la dicha galactosemia. Ésta se caracteriza por causar daños en el hígado, riñones y sistema nervioso central, entre otros sistemas.
Tal y como se verá a continuación, hay varios tipos de galactosemia causadas por el déficit de distintas enzimas.
Es muy importante una detección precoz del problema, y además un buen tratamiento para superar esta enfermedad.

Genética
La Galactosemia sigue un patrón de herencia autosómico recesivo , eso quiere decir que para que el sujeto en cuestión presente la enfermedad, ha de presentar dos copias de un gen anormal.
Los padres son portadores sanos heterocigotos, éstos transmitirán la enfermedad a un 25% de los hijos, al haber heredado éste, el gen alterado de cada uno de sus progenitores. Otro 25% de los hijos no padecerá ni será portador de la enfermedad; y finalmente un 50% de los descendientes serán portadores asintomáticos. Así pues, una persona con un solo alelo, se dice que es “portador” pero no padece la enfermedad, sin embargo, éste puede pasar el gen anormal a sus descendientes, y éstos sí manifestarlo.
Los dos tipos de galactosemia más tratados son: la galactosemia clásica y la galactosemia Duarte. La galactosemia clásica se debe a que el individuo no sintetiza la enzima GALT por lo que éste no puede degradar la galactosa. En la galactosemia Duarte, por contra, sí se sintetiza la enzima GALT, pero de forma deficiente (Véase más adelante).
Generalmente los padres portadores asintomáticos tendrán un 25% de los hijos sanos, un 50% de los hijos portadores asintomáticos y un 25% de los hijos con galactosemia. Gene map locus: 9p13






.jpg)